П/к. Начальная доза препарата Пралуэнт составляет 75 мг, которую вводят 1 раз в 2 нед или 300 мг 1 раз каждые 4 нед (ежемесячно). У пациентов, которым требуется большее снижение концентрации Хс-ЛПНП (>60%), начальная доза препарата Пралуэнт может составлять 150 мг, которую также вводят 1 раз в 2 нед.
Дозу препарата Пралуэнт следует подбирать индивидуально, на основании таких параметров, как исходные значения Хс-ЛПНП, цели терапии и ответ пациента на лечение. Концентрации липидов в крови можно оценивать через 4–8 нед после начала лечения или титрования дозы и проводить соответствующую коррекцию дозы. При необходимости дополнительного снижения концентрации Хс-ЛПНП у пациентов, которым препарат Пралуэнт назначался в дозе 75 мг 1 раз каждые 2 нед или 300 мг 1 раз каждые 4 нед, доза может быть скорректирована до максимальной дозы 150 мг 1 раз каждые 2 нед.
В случае пропуска дозы пациент должен получить инъекцию как можно скорее и затем продолжить лечение по исходной схеме.
Особые группы пациентов
Дети. Безопасность и эффективность применения препарата Пралуэнт у детей в возрасте до 18 лет не установлены.
Пожилой возраст. У пациентов пожилого возраста коррекции дозы препарата Пралуэнт не требуется (см. «Особые указания»).
Печеночная недостаточность. У пациентов с печеночной недостаточностью легкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется.
Данные по применению препарата Пралуэнт у пациентов с печеночной недостаточностью тяжелой степени отсутствуют.
Почечная недостаточность. У пациентов с почечной недостаточностью легкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется. Данные по применению препарата у пациентов с почечной недостаточностью тяжелой степени ограничены.
Масса тела. Не требуется коррекции режима дозирования в зависимости от массы тела пациентов.
Правила введения препарата
Препарат Пралуэнт применяют в виде п/к инъекций, проводимых в области бедра, живота или плеча, с помощью одноразовой предварительно заполненной шприц-ручки или одноразового предварительно заполненного шприца.
Рекомендуется менять места инъекций при проведении каждой инъекции.
При назначении дозы 300 мг препарат вводят в виде двух инъекций по 150 мг в разные места.
Препарат Пралуэнт не следует вводить в области активных кожных заболеваний или повреждений, таких как солнечные ожоги, кожная сыпь, воспаления кожи или кожные инфекции.
Не следует вводить препарат Пралуэнт в то же место, в которое вводились другие лекарственные препараты.
Описание предварительно заполненных одноразовых шприц-ручек и инструкция по их использованию
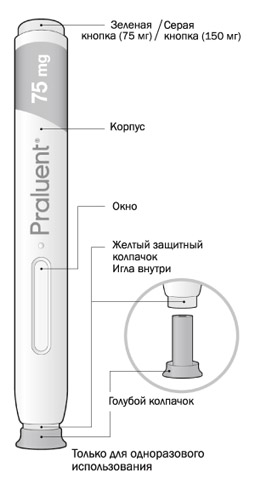
Пралуэнт, 75 мг, раствор для п/к введения в предварительно заполненной шприц-ручке.
Пралуэнт, 150 мг, раствор для п/к введения в предварительно заполненной шприц-ручке.
Инструкция по применению
Пациента необходимо информировать о том, что он должен сохранять эту инструкцию, т.к. она может понадобиться ему снова. Если у пациента появятся вопросы, то ему следует обратиться к лечащему врачу или провизору или позвонить по номеру телефона Представительства АО «Санофи-авентис груп», указанному в инструкции.
Части шприц-ручки указаны на рисунке
Важная информация
- шприц-ручка предназначена только для одноразового использования;
- шприц-ручка с зеленой кнопкой: в 1 мл раствора содержится 75 мг алирокумаба;
- шприц-ручка с серой кнопкой: в 1 мл раствора содержится 150 мг алирокумаба;
- препарат вводится п/к либо самим пациентом, либо другим лицом, ухаживающим за ним;
- шприц-ручку можно использовать только для одноразовой инъекции, и она должна быть утилизирована после применения.
Правила использования
- шприц-ручку следует хранить в недоступном для детей месте;
- перед использованием шприц-ручки следует внимательно прочитать инструкцию;
- необходимо следовать всем указаниям, представленным в инструкции, при каждом использовании шприц-ручки;
- неиспользованные шприц-ручки следует хранить в холодильнике при температуре от 2 до 8 °C.
Запрещено
- трогать желтый защитный колпачок;
- использовать шприц-ручку при протекании или повреждении;
- использовать шприц-ручку при отсутствии голубого колпачка или если он ненадежно закреплен;
- использовать шприц-ручку повторно;
- трясти шприц-ручку;
- замораживать шприц-ручку;
- подвергать шприц-ручку воздействию прямых солнечных лучей.
Этап А. Подготовка к введению
Перед введением препарата пациенту понадобятся:
- шприц-ручка с препаратом Пралуэнт;
- салфетки, смоченные спиртом;
- ватные тампоны или марля;
- контейнер, резистентный к проколам.
1. Проверить этикетку на шприц-ручке:
- убедиться, что взят правильный (нужный пациенту) препарат и правильная (нужная пациенту) доза (шприц-ручка с зеленой кнопкой — для дозы 75 мг/мл и шприц-ручка с серой кнопкой — для дозы 150 мг/мл);
- проверить срок годности. Не использовать по истечении срока годности.
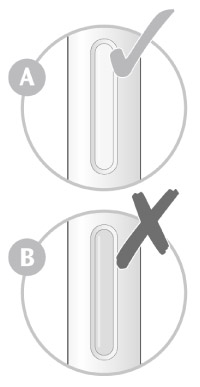
2. Проверить окно:
- проверить, чтобы видимая в окно жидкость была прозрачной и бледно-желтой, без включений. Если имеются изменения цвета и видимые включения, то не следует использовать шприц-ручку (см. рисунок А);
- пациент может увидеть пузырьки воздуха. Это нормальное явление, не влияет на дальнейшее использование препарата;
- не использовать шприц-ручку, если окно желтого цвета и не содержит жидкости (см. рисунок B).
3. Оставить шприц-ручку согреваться при комнатной температуре в течение 30–40 мин:
- не нагревать шприц-ручку, она должна согреться сама;
- использовать шприц-ручку как можно скорее после того, как она нагрелась;
- не класть шприц-ручку обратно в холодильник.
4. Подготовка места введения препарата:
- вымыть руки водой с мылом и вытереть их полотенцем;
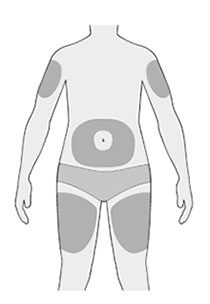
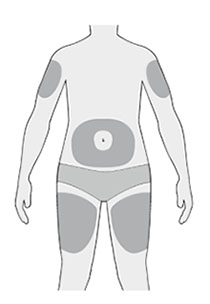
- можно вводить препарат в область бедра, живота (за исключением области 5 см вокруг пупка), наружную поверхность плеча (см. рисунок);
- пациент может стоять или сидеть во время введения препарата;
- продезинфицировать место введения салфеткой, смоченной спиртом;
- не вводить препарат в болезненную, уплотненную, покрасневшую или воспаленную кожу;
- не вводить препарат в места рядом с видимыми венами;
- использовать разные места при каждом введении препарата.
- не вводить препарат в одно и то же место с другими препаратами.
Этап B. Как вводить препарат
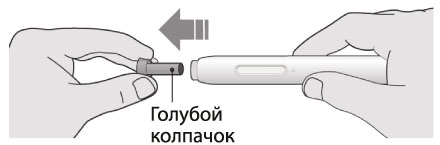
1. После завершения всех пунктов «Этап А. Подготовка к введению» снять голубой колпачок:
- не снимать колпачок до тех пор, пока пациент не будет готов к введению препарата;
- не надевать голубой колпачок обратно.
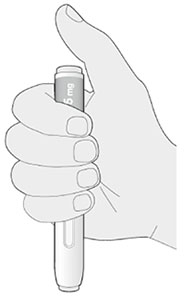
2. Держать шприц-ручку, как показано на рисунке:
- не трогать желтый защитный колпачок;
- убедиться, что видно окно.
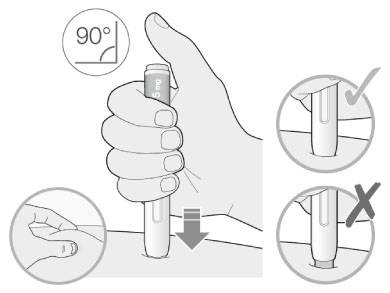
3. Прижать желтый защитный колпачок к коже примерно под углом 90°:
- прижимать шприц-ручку к коже и удерживать ее так, чтобы желтый защитный колпачок стал невидимым (вдавленным в кожу). Шприц-ручка не будет работать, если желтый защитный колпачок не вдавлен в кожу полностью;
- при необходимости следует зажать кожу, чтобы убедиться, что место введения плотное.
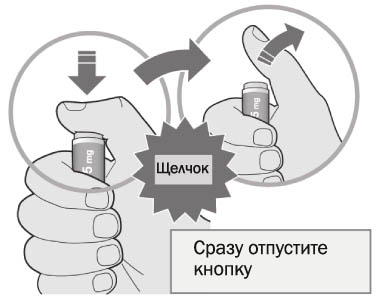
4. Нажать большим пальцем на зеленую (если используется шприц-ручка Пралуэнт, 75 мг) или серую кнопку (если используется шприц-ручка Пралуэнт, 150 мг) и сразу отпустить ее:
- будет слышен щелчок. Введение препарата началось;
- окно начнет желтеть.
5. Продолжать удерживать шприц-ручку плотно прижатой к коже после того, как отпущена кнопка:
- введение препарата может продолжаться до 20 с.
6. Проверить, что окно стало желтого цвета до того, как убрана шприц-ручка с кожи:
- не убирать шприц-ручку с кожи до тех пор, пока все окно не будет желтого цвета;
- введение препарата завершено, после того как все окно будет желтого цвета и будет слышен второй щелчок;
- если окно не стало полностью желтого цвета, не следует вводить самостоятельно вторую дозу без консультации врача.
7. Убрать шприц-ручку с кожи:
- не растирать кожу после введения препарата;
- если пациент видит кровь, необходимо прижать ватный тампон или марлю и удерживать их до тех пор, пока не остановится кровотечение.
8. Утилизировать шприц-ручку и колпачок:
- не надевать голубой колпачок повторно;
- поместить шприц-ручку в контейнер, резистентный к проколам;
- всегда хранить контейнер в недоступном для детей месте.
Описание предварительно заполненных одноразовых шприцев и инструкция по их использованию
Пралуэнт, 75 мг, раствор для п/к введения в предварительно заполненном шприце.
Пралуэнт, 150 мг, раствор для п/к введения в предварительно заполненном шприце.
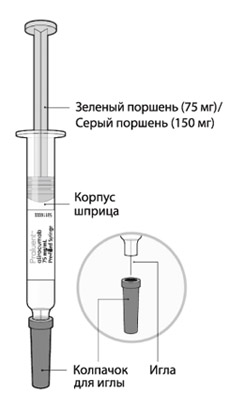
Части шприца показаны на рисунке.
Важная информация
Шприц предназначен для одноразового использования.
- шприц с зеленым поршнем: в 1 мл раствора содержится 75 мг алирокумаба;
- шприц с серым поршнем: в 1 мл раствора содержится 150 мг алирокумаба;
- препарат вводится п/к либо самим пациентом, либо другим лицом, ухаживающим за ним;
- шприц можно использовать только для однократной инъекции, и он должен быть утилизирован после использования.
Правила использования
- шприц следует хранить в недоступном для детей месте;
- перед использованием шприца следует внимательно прочитать инструкцию;
- необходимо следовать всем указаниям, представленным в инструкции, при каждом использовании шприца;
- неиспользованные шприцы следует хранить в холодильнике при температуре от 2 до 8 °C.
Запрещено
- трогать иглу;
- использовать шприц при протекании или повреждении;
- использовать шприц при отсутствии серого колпачка для иглы, или если он ненадежно закреплен;
- использовать шприц повторно;
- трясти шприц;
- замораживать шприц;
- подвергать шприц воздействию прямых солнечных лучей.
Этап А. Подготовка к введению
Перед началом введения пациенту понадобятся:
- шприц с препаратом Пралуэнт;
- салфетки, смоченные спиртом;
- ватные тампоны или марля;
- контейнер, резистентный к проколам.
1. Перед началом следует:
- взять шприц из пакета, удерживая его за корпус.
2. Проверить этикетку на шприце:
- проверить, что взят правильный (нужный пациенту) препарат и правильная (нужная пациенту) доза (шприц с зеленым поршнем — для дозы 75 мг/мл и шприц с серым поршнем — для дозы 150 мг/мл);
- проверить срок годности, не использовать по истечении срока годности;
- проверить, чтобы жидкость в шприце была прозрачной и бледно-желтой, без включений. Если имеются изменения цвета и видимые включения, то не следует использовать шприц;
- проверить, что шприц не открыт и не поврежден.
3. Оставить шприц согреваться при комнатной температуре в течение 30–40 мин:
- не нагревать шприц, он должен согреться сам;
- использовать шприц как можно скорее после того, как он согреется;
- не класть шприц обратно в холодильник.
4. Подготовка места введения:
- вымыть руки водой и мылом и вытереть их полотенцем;
- можно вводить препарат в область бедра, живота (за исключением области 5 см вокруг пупка), наружную поверхность плеча (см. рисунок);
- пациент может стоять или сидеть во время введения препарата;
- продезинфицировать место введения салфеткой, смоченной спиртом;
- не вводить препарат в болезненную, уплотненную, покрасневшую или воспаленную кожу;
- не вводить препарат в места рядом с видимыми венами;
- использовать разные места при каждом введении препарата.
Этап B. Как вводить препарат
1. После завершения всех пунктов «Этап А. Подготовка к введению» снять колпачок для иглы:
- не снимать колпачок до тех пор, пока пациент не будет готов к введению препарата;
- держать шприц за середину корпуса так, чтобы игла была направлена от пациента;
- не прикасаться к поршню;
- пациент может увидеть пузырьки воздуха. Это нормальное явление, не влияет на дальнейшее использование препарата. Не следует избавляться ни от каких пузырьков в шприце до начала инъекции;
- не надевать серый колпачок обратно.
2. При необходимости зажать кожу:
- большим и указательным пальцем зажать кожу так, чтобы получилась складка кожи в месте введения;
- удерживать кожную складку во время проведения всей инъекции.
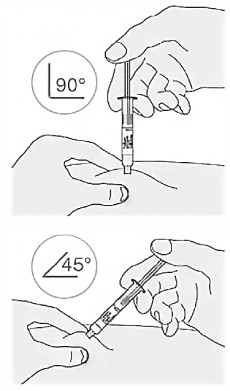
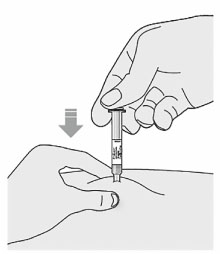
3. Ввести иглу в складку кожи быстрым движением:
- использовать угол в 90° при сжатии 5 см кожи; использовать угол в 45° при сжатии 2 см кожи.
4. Опустить поршень вниз:
- ввести весь раствор медленным и равномерным нажатием на поршень;
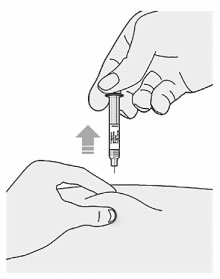
5. Перед тем как удалить иглу, следует убедиться, что шприц пуст:
- не удалять шприц, пока он полностью не опустеет;
- вытаскивать иглу из кожи следует под тем же углом, под каким она была введена;
- не растирать кожу после введения препарата;
- если видна кровь, необходимо прижать ватный тампон или марлю и удерживать их до тех пор, пока не остановится кровотечение.
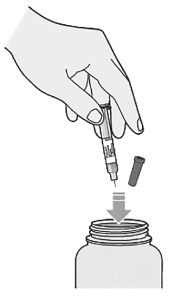
6. Утилизировать шприц и колпачок:
- не надевать серый колпачок повторно;
- не использовать шприц повторно;
- поместить шприц в контейнер, резистентный к проколам;
- всегда хранить контейнер в недоступном для детей месте.
Подготовка и обращение с препаратом
- пациент может вводить препарат Пралуэнт самостоятельно, или препарат может быть введен лицом, ухаживающим за пациентом, после предоставления им медицинскими работниками информации по правильной технике проведения п/к инъекций;
- препараты для парентерального введения должны осматриваться визуально перед введением на предмет наличия видимых частиц и изменения цвета. Если раствор имеет измененный цвет или видимые частицы, его не следует использовать;
- перед использованием предварительно заполненных шприц-ручек и предварительно заполненных шприцев следует согреть их до комнатной температуры;
- препарат Пралуэнт следует использовать как можно скорее, после того как он согреется. Препарат может храниться не более 24 ч при температуре не выше 25 °C;
- после использования шприц-ручки и шприцы следует поместить в резистентный к проколам контейнер и утилизировать согласно местным (государственным) требованиям;
- не использовать контейнер повторно;
- всегда следует хранить контейнер в местах, недоступных для детей;
- пациенты и ухаживающие за ними лица должны получить руководство по надлежащей технике п/к введения, включая сведения по асептике, и по тому, как правильно использовать предварительно заполненные шприц-ручки и предварительно заполненные шприцы;
- пациентов и ухаживающих за ними лиц следует информировать о том, что перед первым введением препарата Пралуэнт они должны внимательно прочитать инструкцию по применению препарата;
- пациентов и ухаживающих за ними лиц следует предупредить, что запрещено повторное использование шприц-ручек или шприцев, и они должны быть проинструктированы, как безопасно утилизировать их после использования.